|
A fatal hereditary disease for Shibas
|
In the year 2000 a Shiba was diagnosed for the first time in Japan with a rare hereditary disease that was known in different breeds but until then had not occurred in Shibas. This concerns a so-called storage disorder (metabolic disease) by the name of GM1 gangliosidosis which is not curable and is fatal within one and a half years.
The disease was detected by a group of veterinary physicians working with Dr. Osamu Yamato at the Hokkaido University in Sapporo. Within a short time these scientists had discovered the cause of this disease in Shibas (a gene mutation) and since then have developed a new diagnostic method. They assume that the disease is already widely spread amongst Shibas in Japan and are concerned that it might have reached America and Europe also. Even though there is currently no concrete cause for anxiety outside Japan, Shiba breeders should be informed about this new disease.
 What is gangliosidosis?
What is gangliosidosis?
Gangliosidosis belongs to a group of hereditary diseases known as "lysosomal storage diseases".
Lysosomes are specific structures inside the cell in which, as in a "sewage plant", many substances are degraded or altered. Molecules carrying out this degrading or altering are called enzymes. With a lysosomal storage disorder, substances that due to an enzyme deficiency cannot be processed are stored in the lysosomes. In the case of gangliosidosis, gangliosides (fat-sugar compounds) build up in the brain cells. Thereby vital cell functions in the brain are mutilated resulting in severe disorder symptoms.
Gangliosidosis occurs in two different forms. The GM1 gangliosidosis is caused by an inherited deficiency of the beta-galactosidase enzyme. In this form the neurological symptoms begin slightly later (approx. at 4 months) and proceed more slowly. With GM2 gangliosidosis the beta-hexosaminidase enzyme is missing, the disease pattern normally appears earlier and aggravates quicker. Even though both forms of gangliosidosis have similar symptoms, they are evoked by completely different defects of two specific lysosomal enzymes. These genetic defects known as mutations are due to a modification in the genetic code.
What makes both of these diseases pernicious is that they are inherited covertly (autosomal recessive). With this transmission, the illness only breaks out when both formations of a critical gene are defective (as opposed to autosomal dominant inheritance where the illness breaks out when only one critical gene is defective.) The heredity course of gangliosidosis follows the classical laws of genetics. An individual always inherits a gene copy from the dam and one from the sire. A recessive heredity course means that dogs carrying just one copy of the defective gene (i.e. they are heterozygote) are clinically healthy but are carriers. Carriers themselves will never get this disease but there is a 50% chance that they will pass the defective gene on to their offspring. Only if two copies of the defective gene (from dam and sire) exist, i.e. the puppy is homzygous, will the disease break out. If carriers are mated, 25% of their offspring will statistically get gangliosidosis, 50% will be carriers and 25% will be free of the defective gene.
Even when mating hereditarily sound dogs and carriers caution should be exercised. There is no chance of offspring being affected by gangliosidosis but with a probability of 50% carriers will be produced. Thereby the defective gene can be spread unwittingly in the dog population.
Who can contract gangliosidosis?
As well as humans, cats, sheep, calves and dogs have also contracted gangliosidosis. The course of the disease with dogs has the most similarity to that with humans and has therefore been researched intensively.
With dogs the GM1 gangliosidosis has been verified with mix-breed
Beagles (1976), English Springer Spaniels (1983), Portuguese Water Dogs (1988), Alaskan Huskies (1998) and a mongrel of unknown origin (2000). Due to a veterinary thesis at the University of Gießen (Germany) this disease in Alaskan Huskies is well researched.
[1]
[2]
GM2 gangliosidosis has been recorded with the German Short-haired
Pointer (1967/1989), the Japanese Spaniel (Chin) (1985) and with the Golden Retriever (2002). The German Short-haired Pointer showed neurological symptoms at the age of 6 to 9 months in the form of visual problems, incoordination, abnormal behaviour and a stiff gait. With the Chin the disease pattern was similar but the dog was already two years old. The Golden Retriever was suffering from the so-called Sandhoff variant of GM2 gangliosidosis.
How does the veterinarian recognise GM1 gangliosidosis?
Diagnoses are performed by means of clinical symptoms, laboratory tests and pathological abnormalities of the brain. Molecular tests for this disorder are as yet rather complicated. Currently in Japan the group around Dr. Yamato is developing new diagnostic methods on the basis of the disease patterns in Shibas.
[3]
[4]
[5]
The disease pattern with Japanese Shibas
For the first time in 1999/2000 the GM1 gangliosidosis was diagnosed in a 6-month-old female, purebred Shiba which from the age of one month had suffered from motoric dysfunctions and showed symptoms of a brain disorder including motoric coordination problems (ataxia, dysmetria) and head tremors. A sibling with a beta-galactosidase deficiency in the brain and visceral organs had died immediately after birth. The sire and dam of the puppies had half of the normal level of beta-galactosidase activity and were heterozygotes. The disease pattern suggested an autosomal recessive hereditary disease.
[6]
[7]
Two years later the diagnosis was confirmed. A homozygous recessive mutation was identified to be the direct cause of GM1 gangliosidosis with Shibas. As a consequence the cytosine base in position 1647 of the coding region of the beta-galactosidase gene has been deleted. This was a novel mutation in canine GM1 gangliosidosis.
[4]
[8]
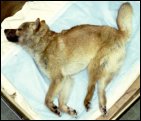
A year further on researchers working with Dr. Yamato published a study describing the disease pattern with affected Shibas as follows. The disorder broke out at an age of 5 to 6 months whereby the dogs displayed progressive neurological symptoms including loss of balance, sporadic paralyses, ataxia, dysmetria and intention tremor of the head. At the age of 10 months the dogs were unable to stand. Between 9 to 12 months corneal clouding, visual defects, general muscle paralysis, emotional disorders and a tendency to lethargy were observed. The dogs became lethargic from 13 months of age. Their life expectancy was not more than 15 months.
[9]
The following information and pictures have been provided by courtesy of Dr. Yamato, Hokkaido University in Sapporo, Japan. They illustrate the course of the disease showing different affected Shibas.
| Age (months) |
Clinical signs |
|
<5
|
No clinical or neurological signs |
|
5-6
|
Loss of balance, Lameness (intermittent), Ataxia (mild to moderate), Dysmetria (mainly hypermetria), Head tremor (intention tremor) |
|
7-8
|
Ataxia (severe), Toppling gait, Exaggeratedly startled response |
|
9-10
|
Atactic abasia, Astasia, Corneal clouding, Visual defect, Muscle rigospasticity in limbs and crest, Emotional disorder |
|
11-12
|
Generalised muscle rigospasticity, Tonic spasm, Tendency to be lethargic, Unresponsive to sounds, Weight loss |
|
13<
|
Lethargy, Death (mainly after 14 months) |
1 month

|
4 months

|
6 months

|
9 months

|
11 months

|
13 months
|
References
Note: On request the Shiba Klub of the Czech Republic will provide abstracts of the following articles as well as an abstract of the thesis on the disease in the Alaskan Husky in Word format.
Article no. 9 ("Clinical and clinico-pathologic characteristics ...") can be provided as full text in PDF format. Please send us an
e-mail.
|
[1] |
Gundi Müller: Die GM1-Gangliosidose beim Alaskan Husky unter besonderer Berücksichtigung der neuropathologischen Veränderungen, PhD (Vet.) thesis, University of Gießen (Germany) 2001.
|
|
[2] |
Robert Kreutzer, Tosso Leeb, Gundi Müller, Andreas Moritz, Wolfgang Baumgärtner: A duplication in the canine β-galactosidase gene GLB1 causes exon skipping and GM1-gangliosidosis in Alaskan Huskies, Genetics 170 (2005), pp. 1857-1861.
|
|
[3] |
Satoh H., Yamato O., Asano T., Yamasaki M., Maede Y.: Increased concentration of GM1-ganglioside in cerebrospinal fluid in dogs with GM1- and GM2-gangliosidoses and its clinical application for diagnosis, Journal of Veterinary Diagnostic Investigation 16 (2004), pp. 223-226.
|
|
[4] |
Yamato O., Kobayashi A., Satoh H., Endoh D., Shoda T., Masuoka Y., Hatakeyama A., Jo E. O., Asano T., Yonemura M., Yamasaki M., Maede Y.: Comparison of polymerase chain reaction-restriction fragment length polymorphism assay and enzyme assay for diagnosis of G(M1)-gangliosidosis in Shiba dogs, Journal of Veterinary Diagnostic Investigation 16 (2004), pp. 299-304.
|
|
[5] |
Yamato O., Jo E. O., Shoda T., Yamasaki M., Maede Y.: Rapid and simple mutation screening of G(M1) gangliosidosis in Shiba dogs by direct amplification of deoxyribonucleic acid from various forms of canine whole-blood specimens, Journal of Veterinary Diagnostic Investigation 16 (2004), pp. 469-472.
|
|
[6] |
Eriko Hayashida: Pathological Studies of GM1 Gangliosidosis in a Shiba Dog, Japanese Journal of Veterinary Medicine 47 (1-2) (1999), pp. 87-88.
Yamato O., Ochiai K., Masuoka Y., Hayashida E., Tajima M., Omae S., Iijima M., Umemura T., Maede Y.: GM1 gangliosidosis in shiba dogs, The Veterinary Record 146 (2000), pp. 493-496.
|
|
[7] |
Tanaka H., Nakayama M., Sugiyama Y., Yamato O.: GM1 gangliosidosis diagnosed on the basis of assay of leukocytic lysosomal-enzyme activity in a Shiba-breed dog, Journal of the Japanese Veterinary Medical Association 54 (2001), pp. 625-627 (in Japanese with English summary).
|
|
[8] |
Yamato O., Endoh D., Kobayashi A., Masuoka Y., Yonemura M., Hatakeyama A., Satoh H., Tajima M., Yamasaki M., Maede Y.: A novel mutation in the gene for canine acid β-galactosidase that causes GM1-gangliosidosis in Shiba dogs,
Journal of Inherited Metabolic Disease 25 (2002), pp. 525-526.
|
|
[9] |
Yamato O., Masuoka Y., Yonemura M., Hatakeyama A., Satoh H., Kobayashi A., Nakayama M., Asano T., Shoda T., Yamasaki M., Ochiai K., Umemura T., Maede Y.: Clinical and clinico-pathologic characteristics of Shiba dogs with a deficiency of lysosomal acid β-galactosidase: a canine model of human GM1 gangliosidosis, Journal of Veterinary Medical Science 65 (2003), pp. 213-217.
|
© 2005 Holger Funk

;)
;)
;)
;)
;)
;)